Marker Genes Calculation and Visualization
Loading Packages
[ ]:
import warnings
warnings.filterwarnings('ignore')
import scanpy as sc
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
from sklearn.preprocessing import MinMaxScaler
import sys
sys.path.append(r'../../../')
from SpatialCOC.preprocess import preprocessing
from SpatialCOC.utils import fix_seed, rotate_spatial_coordinates, reorder_categories, replace_extreme_values
fix_seed(2024)
Loading Data
[9]:
slice_id = 4 ## 1, 2, 3, 4
slice_name = ['ATAC', 'H3K4me3', 'H3K27ac', 'H3K27me3']
adata_modality_1 = sc.read_h5ad(f"../../../Data/Mouse_Brain_{slice_name[slice_id-1]}/adata_RNA.h5ad")
adata_modality_2 = sc.read_h5ad(f"../../../Data/Mouse_Brain_{slice_name[slice_id-1]}/adata_peaks_normalized.h5ad")
adata_modality_1, adata_modality_2 = preprocessing(adata_modality_1, adata_modality_2, 'Spatial-epigenome-transcriptome')
adata_results = sc.read_h5ad(f"../../Mouse_Brain_{slice_name[slice_id-1]}.h5ad")
Spatial-epigenome-transcriptome data preprocessing have done!
Dimensions after preprocessed adata_modal_1: (9732, 3000)
Dimensions after preprocessing adata_modal_2: (9732, 70470)
Calculating Marker Genes
[10]:
adata_modality_1.obs['SpaKnit'] = adata_results.obs['SpaKnit']
## calculate the marker genes
sc.tl.dendrogram(adata_modality_1, groupby='SpaKnit')
sc.tl.rank_genes_groups(adata_modality_1, groupby='SpaKnit', use_raw=False)
## store the results
rank_genes = sc.get.rank_genes_groups_df(adata_modality_1, group=None)
rank_genes.to_excel(f"{slice_name[slice_id-1]}/Marker_Genes.xlsx", index=True)
WARNING: Default of the method has been changed to 't-test' from 't-test_overestim_var'
Ploting Spatial Distribution of Marker Genes
[40]:
## Define Marker gene list
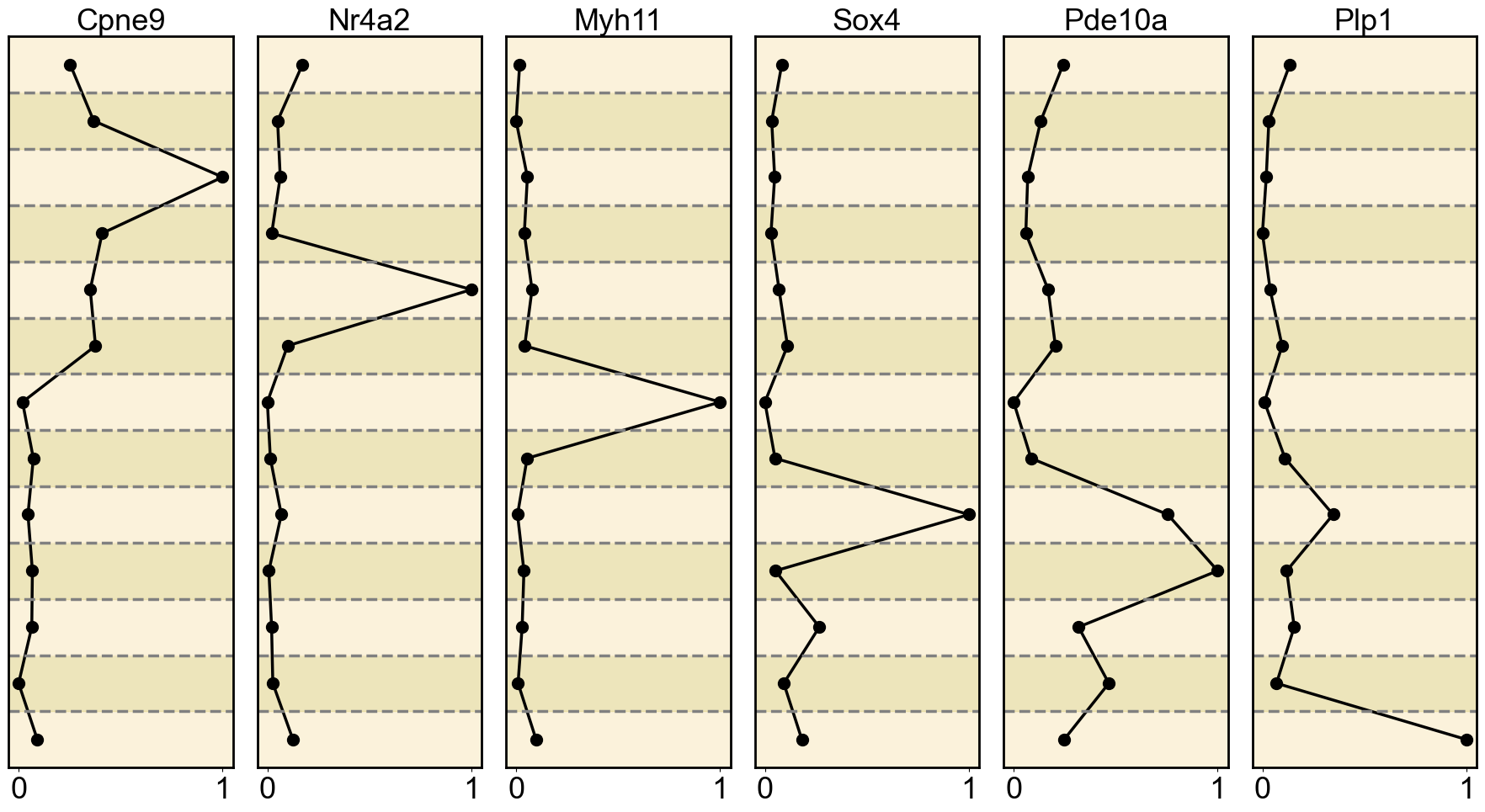
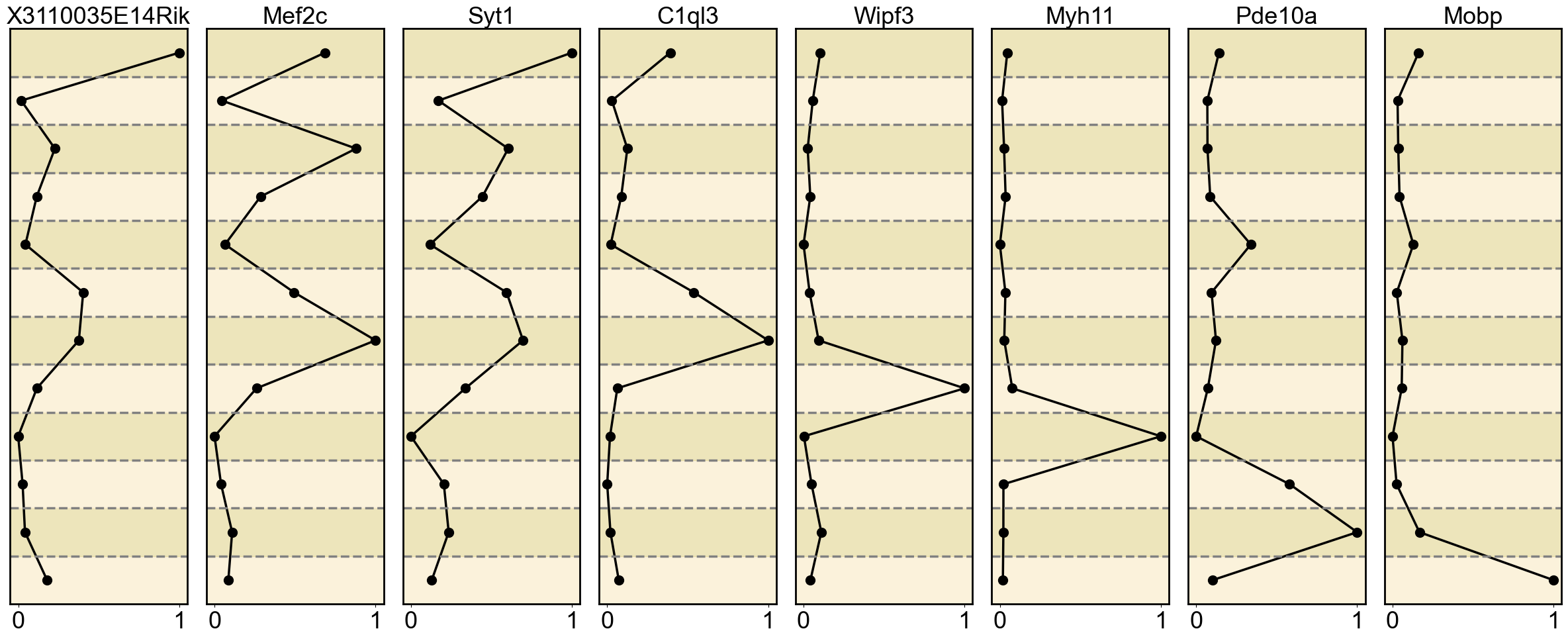
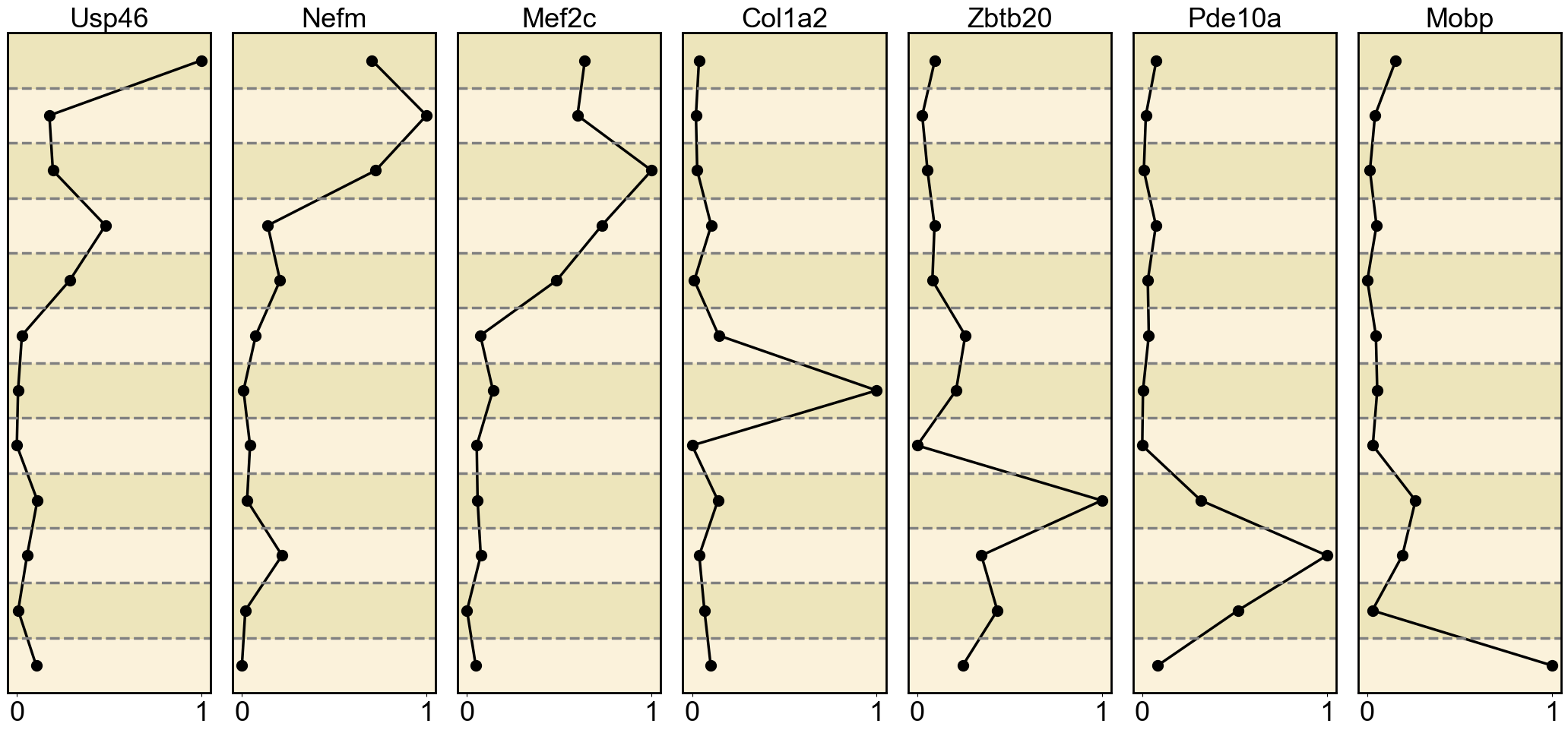
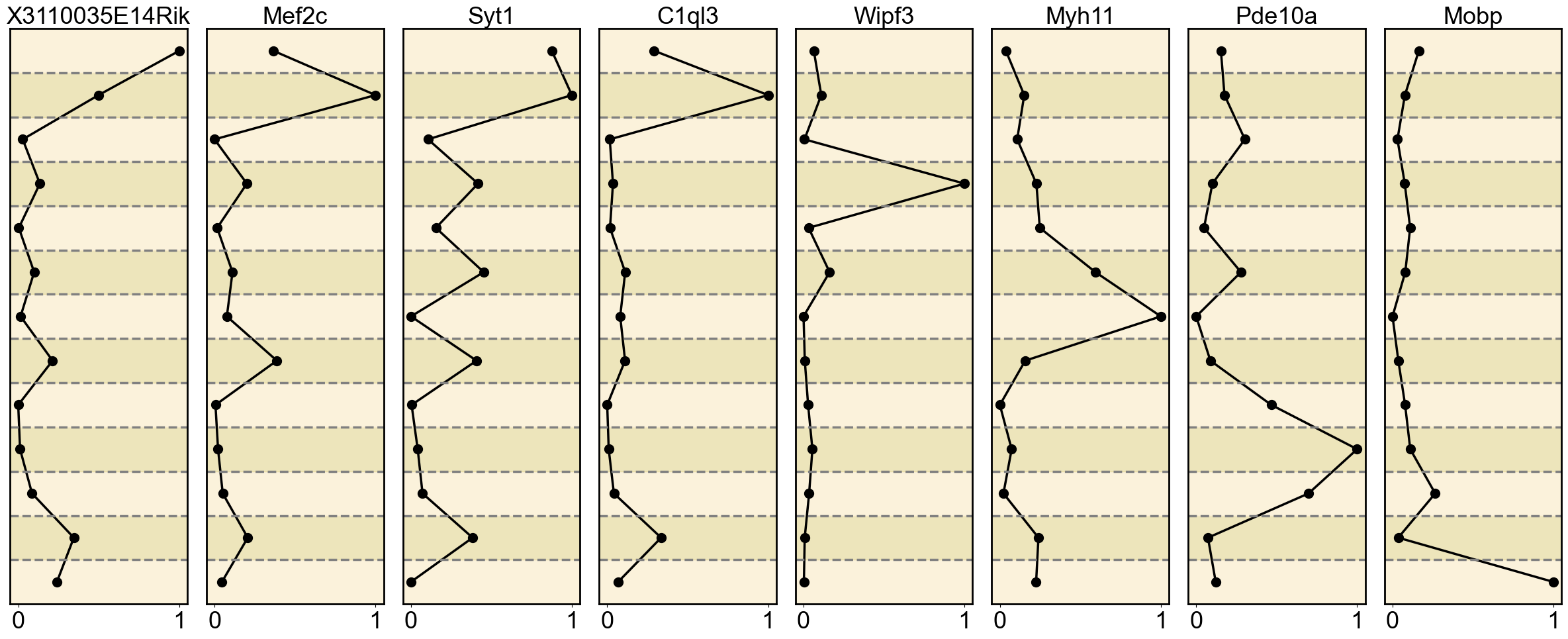
gene_list = {
'ATAC': ['Cpne9', 'Nr4a2', 'Myh11', 'Sox4', 'Pde10a', 'Plp1'],
'H3K4me3': ['X3110035E14Rik', 'Mef2c', 'Syt1', 'C1ql3', 'Wipf3', 'Myh11', 'Pde10a', 'Mobp'],
'H3K27ac': ['Usp46', 'Nefm', 'Mef2c', 'Col1a2', 'Zbtb20', 'Pde10a', 'Mobp'],
'H3K27me3': ['X3110035E14Rik', 'Mef2c', 'Syt1', 'C1ql3', 'Wipf3', 'Myh11', 'Pde10a', 'Mobp']
}
for slice_id in range(0, 4):
current_slice = slice_name[slice_id]
print(f"Processing slice: {current_slice}")
# Read data
adata_modality_1 = sc.read_h5ad(f"../../../Data/Mouse_Brain_{current_slice}/adata_RNA.h5ad")
# Data preprocessing
sc.pp.filter_genes(adata_modality_1, min_cells=10)
sc.pp.filter_cells(adata_modality_1, min_genes=200)
sc.pp.highly_variable_genes(adata_modality_1, flavor="seurat_v3", n_top_genes=3000)
sc.pp.normalize_total(adata_modality_1, target_sum=1e4)
# Get the marker genes for the current slice
current_genes = gene_list[current_slice]
num_genes = len(current_genes)
# Set plotting parameters
plt.rcParams['font.size'] = 18
plt.rcParams['font.sans-serif'] = 'Arial'
# Create subplots
fig, axs = plt.subplots(1, num_genes, figsize=(num_genes * 4, 4), squeeze=False)
# Determine rotation angle based on slice_id
rotation_angle = 0
if slice_id == 1 or slice_id == 2: # Rotate 90 degrees clockwise for the second and third slices
rotation_angle = 90
elif slice_id == 3: # Rotate 270 degrees clockwise for the fourth slice
rotation_angle = 270
if rotation_angle != 0:
original_spatial = adata_modality_1.obsm['spatial'].copy()
# Rotate coordinates
adata_modality_1.obsm['spatial'] = rotate_spatial_coordinates(original_spatial, rotation_angle)
# Iterate over each gene and plot
for i, gene in enumerate(current_genes):
try:
expression = adata_modality_1[:, gene].X.toarray().flatten()
# Replace extreme values
adata_modality_1[:, gene].X = replace_extreme_values(expression, n=0.01)
# Plot spatial expression
sc.pl.embedding(adata_modality_1, basis='spatial', color=gene, ax=axs[0, i], s=50, colorbar_loc=None, show=False, cmap='YlGnBu')
# Remove spines and axis labels
for spine in axs[0, i].spines.values():
spine.set_visible(False)
axs[0, i].set_xlabel('')
axs[0, i].set_ylabel('')
axs[0, i].set_title('')
if slice_id == 3:
axs[0, i].invert_xaxis()
except KeyError:
print(f"Gene {gene} not found in {current_slice} data.")
continue
# Adjust layout
plt.subplots_adjust(wspace=0, hspace=0)
plt.tight_layout()
# Save figures
plt.savefig(f'{slice_name[slice_id]}/Marker_Genes_Spatial_{slice_name[slice_id]}.png', dpi=500)
plt.savefig(f'{slice_name[slice_id]}/Marker_Genes_Spatial_{slice_name[slice_id]}.eps')
plt.show()
Processing slice: ATAC
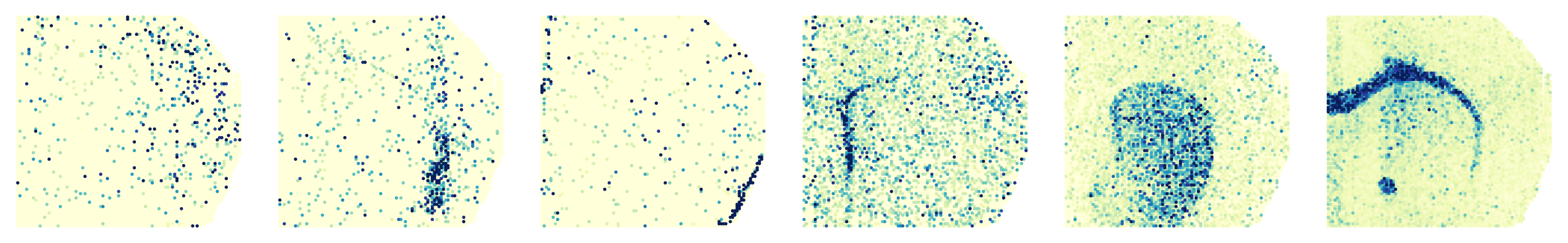
Processing slice: H3K4me3
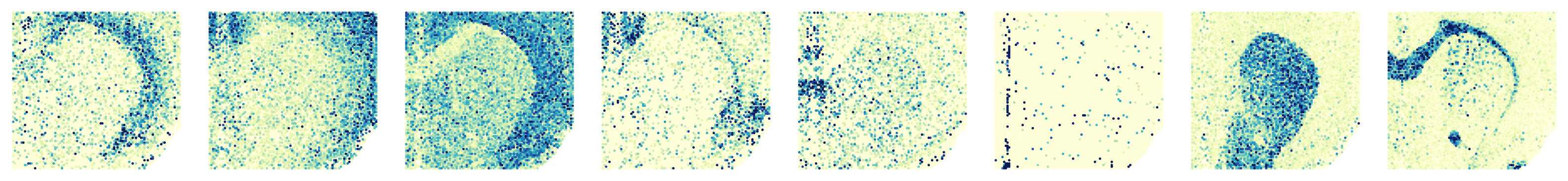
Processing slice: H3K27ac
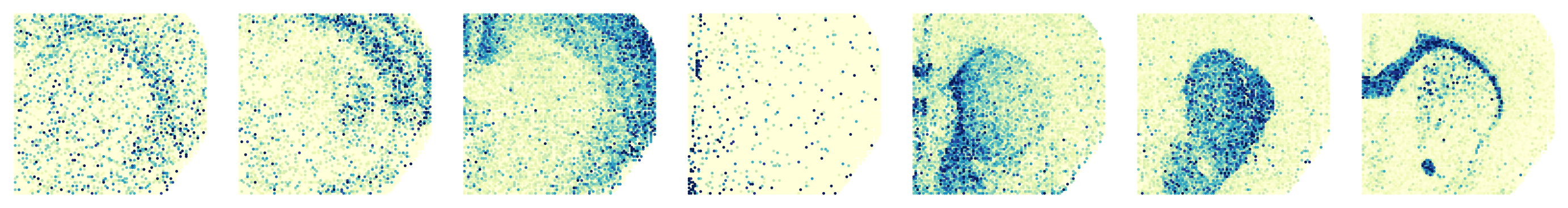
Processing slice: H3K27me3
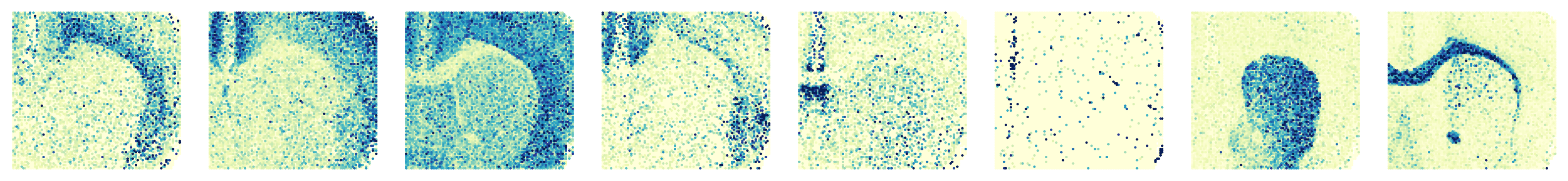
Ploting the Marker genes’ Expression
[146]:
import scanpy as sc
import pandas as pd
import matplotlib.pyplot as plt
import numpy as np
from sklearn.preprocessing import MinMaxScaler
# Define the new orders for each modality
new_orders = {
'ATAC': ["3", "2", "9", "7", "1", "10", "4", "8", "11", "0", "12", "5", "6"],
'H3K4me3': ["4", "2", "3", "7", "10", "5", "8", "11", "9", "1", "0", "6"],
'H3K27ac': ["2", "7", "3", "4", "10", "5", "9", "8", "11", "0", "1", "6"],
'H3K27me3': ["3", "8", "9", "11", "2", "12", "4", "1", "6", "0", "10", "5", "7"]
}
# Define the colors for the background
colors = ['#faedcd', '#e6db9e']
# Iterate over each slice_id
for slice_id in range(4):
current_slice = slice_name[slice_id]
# Load the data
adata_modality_1 = sc.read_h5ad(f"../../../Data/Mouse_Brain_{current_slice}/adata_RNA.h5ad")
adata_results = sc.read_h5ad(f"../../Mouse_Brain_{current_slice}.h5ad")
# Reorder categories
reorder_categories(adata_results, 'SpaKnit', new_orders[current_slice])
# Set the figure size and subplot layout
fig, axes = plt.subplots(nrows=1, ncols=len(gene_list[current_slice]), figsize=(len(gene_list[current_slice]) * 3, 10))
# Iterate over the gene list and create a subplot for each gene
for i, gene in enumerate(gene_list[current_slice]):
# Extract the expression data for the target gene
gene_expression = adata_modality_1[:, gene].X.toarray()
# Convert the expression data to a pandas Series
gene_expression_series = pd.Series(gene_expression.flatten(), index=adata_modality_1.obs.index)
# Extract cell type labels
cell_types = adata_results.obs['SpaKnit']
# Merge gene expression data and cell type labels into a DataFrame
df = pd.DataFrame({'gene_expression': gene_expression_series, 'cell_type': cell_types})
# Group by cell type and calculate the mean gene expression for each group
mean_expression_by_cell_type = df.groupby('cell_type')['gene_expression'].mean()
# Normalize the mean expression values to the range [0, 1]
scaler = MinMaxScaler()
normalized_expression = scaler.fit_transform(mean_expression_by_cell_type.values.reshape(-1, 1)).flatten()
# Plot the normalized line chart
ax = axes[i] # Get the current subplot axis object
ax.plot(normalized_expression, mean_expression_by_cell_type.index, marker='o', color='black', linewidth=2.5, markersize=10)
ax.set_yticks([]) # Remove y-axis labels
ax.set_ylim([-0.5, len(new_orders[current_slice])-0.5]) # Set y-axis limits
ax.invert_yaxis() # Invert the y-axis
ax.set_xlim(-0.05, 1.05) # Set x-axis limits
ax.set_xticks([0, 1]) # Display only 0 and 1 on the x-axis
ax.tick_params(axis='x', labelsize=26)
ax.set_title(gene, fontsize=26)
for spine in ax.spines.values():
spine.set_linewidth(2) # Set the border line width to 2
y_min, y_max = ax.get_ylim()
num_intervals = len(new_orders[current_slice])
y_intervals = np.linspace(y_min, y_max, num_intervals + 1)
for j in range(num_intervals):
lower_bound = y_intervals[j]
upper_bound = y_intervals[j + 1]
# Select color using modulo operation
color = colors[j % 2]
ax.axhspan(lower_bound, upper_bound, color=color, alpha=0.7)
# Draw a gray dashed line at the upper boundary of each interval
if j < num_intervals - 1: # Avoid drawing at the upper boundary of the last interval
ax.axhline(upper_bound, color='gray', linestyle='--', linewidth=2.5, alpha=1)
plt.tight_layout()
plt.savefig(f'{current_slice}/Marker_Gene_Expression_{current_slice}.png', dpi=500)
plt.savefig(f'{current_slice}/Marker_Gene_Expression_{current_slice}.eps')
plt.show()
The PostScript backend does not support transparency; partially transparent artists will be rendered opaque.
The PostScript backend does not support transparency; partially transparent artists will be rendered opaque.
The PostScript backend does not support transparency; partially transparent artists will be rendered opaque.
The PostScript backend does not support transparency; partially transparent artists will be rendered opaque.
[ ]:
import scanpy as sc
import pandas as pd
import numpy as np
from openpyxl import Workbook
from openpyxl.utils.dataframe import dataframe_to_rows
import warnings
warnings.filterwarnings('ignore')
# Define marker genes for each modality
gene_list = {
'ATAC': ['Cpne9', 'Nr4a2', 'Myh11', 'Sox4', 'Pde10a', 'Plp1'],
'H3K4me3': ['X3110035E14Rik', 'Mef2c', 'Syt1', 'C1ql3', 'Wipf3', 'Myh11', 'Pde10a', 'Mobp'],
'H3K27ac': ['Usp46', 'Nefm', 'Mef2c', 'Col1a2', 'Zbtb20', 'Pde10a', 'Mobp'],
'H3K27me3': ['X3110035E14Rik', 'Mef2c', 'Syt1', 'C1ql3', 'Wipf3', 'Myh11', 'Pde10a', 'Mobp']
}
# Define slice names
slice_name = ['ATAC', 'H3K4me3', 'H3K27ac', 'H3K27me3']
# Define new cluster orders for each modality
new_orders = {
'ATAC': ["3", "2", "9", "7", "1", "10", "4", "8", "11", "0", "12", "5", "6"],
'H3K4me3': ["4", "2", "3", "7", "10", "5", "8", "11", "9", "1", "0", "6"],
'H3K27ac': ["2", "7", "3", "4", "10", "5", "9", "8", "11", "0", "1", "6"],
'H3K27me3': ["3", "8", "9", "11", "2", "12", "4", "1", "6", "0", "10", "5", "7"]
}
# Create Excel file with multiple sheets
with pd.ExcelWriter('Marker_Gene_Data.xlsx', engine='openpyxl') as writer:
for slice_id in range(4):
current_slice = slice_name[slice_id]
print(f"Processing slice: {current_slice}")
# Load data files
try:
adata_modality_1 = sc.read_h5ad(f"../../../Data/Mouse_Brain_{current_slice}/adata_RNA.h5ad")
adata_results = sc.read_h5ad(f"../../Mouse_Brain_{current_slice}.h5ad")
except FileNotFoundError as e:
print(f"File not found error: {e}")
continue
# Preprocess the RNA data
sc.pp.filter_genes(adata_modality_1, min_cells=10)
sc.pp.filter_cells(adata_modality_1, min_genes=200)
sc.pp.highly_variable_genes(adata_modality_1, flavor="seurat_v3", n_top_genes=3000)
sc.pp.normalize_total(adata_modality_1, target_sum=1e4)
# ====== Extract spatial coordinates and gene expression ======
print(f" Extracting spatial coordinates and gene expression...")
# Get spatial coordinates
spatial_coords = adata_modality_1.obsm['spatial']
# Create DataFrame for spatial data
spatial_data = pd.DataFrame({
'X_coordinate': spatial_coords[:, 0],
'Y_coordinate': spatial_coords[:, 1]
})
# Add gene expression data
for gene in gene_list[current_slice]:
try:
# Get expression values
expression = adata_modality_1[:, gene].X
if hasattr(expression, 'toarray'):
expression = expression.toarray().flatten()
else:
expression = expression.flatten()
# Add to spatial data
spatial_data[f'{gene}_Expression'] = expression
except KeyError:
print(f" Warning: Gene {gene} not found in {current_slice} dataset")
continue
# Save spatial data to Excel sheet
spatial_sheet_name = f"{current_slice}_Spatial"
if len(spatial_sheet_name) > 31:
spatial_sheet_name = spatial_sheet_name[:31]
spatial_data.to_excel(writer, sheet_name=spatial_sheet_name, index=False)
print(f" Saved spatial data to sheet: {spatial_sheet_name}")
# ====== Extract cluster-specific mean expression ======
print(f" Extracting cluster-specific mean expression...")
# Reorder clusters if needed
try:
if 'SpatialCOC' in adata_results.obs.columns:
# Reorder SpatialCOC clusters
adata_results.obs['SpatialCOC'] = pd.Categorical(
adata_results.obs['SpatialCOC'],
categories=new_orders[current_slice],
ordered=True
)
except Exception as e:
print(f" Warning: Could not reorder clusters: {e}")
# Prepare cluster data
cluster_data_list = []
clusters_available = []
if 'SpatialCOC' in adata_results.obs.columns:
clusters_available = sorted(adata_results.obs['SpatialCOC'].unique())
# Calculate mean expression for each cluster
for cluster in clusters_available:
# Get cells in this cluster
cluster_cells = adata_results.obs[adata_results.obs['SpatialCOC'] == cluster].index
# Calculate mean expression for each gene in this cluster
cluster_expr_data = {}
cluster_expr_data['Cluster'] = cluster
for gene in gene_list[current_slice]:
try:
# Get expression for cells in this cluster
gene_expression = adata_modality_1[cluster_cells, gene].X
if hasattr(gene_expression, 'toarray'):
gene_expression = gene_expression.toarray().flatten()
else:
gene_expression = gene_expression.flatten()
# Calculate mean expression
mean_expr = np.mean(gene_expression)
cluster_expr_data[f'{gene}_Mean_Expression'] = mean_expr
except KeyError:
cluster_expr_data[f'{gene}_Mean_Expression'] = np.nan
continue
except Exception as e:
print(f" Error processing gene {gene} for cluster {cluster}: {e}")
cluster_expr_data[f'{gene}_Mean_Expression'] = np.nan
continue
cluster_data_list.append(cluster_expr_data)
# Create DataFrame from cluster data
if cluster_data_list:
cluster_data = pd.DataFrame(cluster_data_list)
# Sort clusters according to specified order
try:
cluster_data['Cluster'] = pd.Categorical(
cluster_data['Cluster'],
categories=new_orders[current_slice],
ordered=True
)
cluster_data = cluster_data.sort_values('Cluster')
except:
# If reordering fails, keep original order
pass
# Set cluster as index
cluster_data.set_index('Cluster', inplace=True)
# Normalize expression values to 0-1 range
print(f" Normalizing expression values to 0-1 range...")
gene_columns = [col for col in cluster_data.columns if col.endswith('_Mean_Expression')]
for col in gene_columns:
# Get data for normalization
data = cluster_data[col].values
# Skip if all values are NaN
if not np.all(np.isnan(data)):
# Find min and max values
min_val = np.nanmin(data)
max_val = np.nanmax(data)
# Perform 0-1 normalization
if max_val != min_val:
# Standard 0-1 normalization
normalized_data = (data - min_val) / (max_val - min_val)
else:
# If all values are the same
if max_val == 0:
normalized_data = np.zeros_like(data)
else:
normalized_data = np.ones_like(data)
# Replace original data with normalized data
cluster_data[col] = normalized_data
# Rename column to indicate normalization
new_col_name = col.replace('_Mean_Expression', '_Normalized')
cluster_data.rename(columns={col: new_col_name}, inplace=True)
# Save cluster data to Excel sheet
cluster_sheet_name = f"{current_slice}_Cluster"
if len(cluster_sheet_name) > 31:
cluster_sheet_name = cluster_sheet_name[:31]
cluster_data.to_excel(writer, sheet_name=cluster_sheet_name, index=True)
print(f" Saved cluster expression data to sheet: {cluster_sheet_name}")
else:
print(f" No cluster data available (SpatialCOC clustering not found)")
print("Processing complete! Results saved to: Marker_Gene_Data.xlsx")